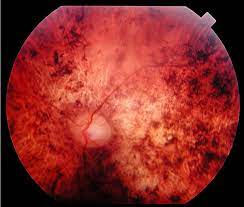
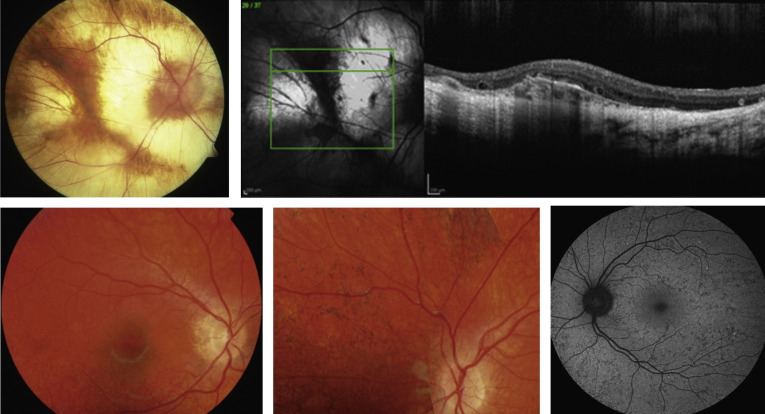
En RP inverso, la pérdida visual se producirá primero en la vista central. Los pines que desaparecen primero son esenciales para leer y detallar. Normalmente, la lectura se está deteriorando temprano.
Esta es una afección hereditaria que se caracteriza por pérdida auditiva y pérdida progresiva de la visión. La pérdida de la visión se debe a la Retinitis Pigmentosa (RP), una afección degenerativa de la retina, y generalmente aparece durante la adolescencia o la adultez temprana. El equilibrio también puede verse afectado. Los síntomas varían de persona a persona y progresan a diferentes velocidades. Los investigadores están estudiando las causas y los posibles tratamientos para el síndrome de Usher y otras enfermedades degenerativas de la retina. Se ha realizado un excelente progreso en la investigación recientemente y varios ensayos clínicos están en progreso.
Hay al menos tres formas diferentes de síndrome de Usher. Las personas con síndrome de Usher tipo 1 (USH1) suelen nacer con pérdida auditiva severa y problemas de equilibrio. Los primeros signos de RP - ceguera nocturna y pérdida de la visión periférica - generalmente aparecen en la adolescencia temprana.
En el síndrome de Usher tipo 2 (USH2), los recién nacidos tienen una discapacidad auditiva de moderada a severa. Los síntomas de RP generalmente comienzan poco después de la adolescencia. Los problemas visuales progresan menos rápidamente que en Usher Tipo 1 y la pérdida auditiva generalmente permanece estable.
En el tercer tipo más raro del síndrome de Usher (USH3), los niños generalmente nacen con un deterioro leve o leve de la audición. Su pérdida de audición y visión es progresiva, comenzando alrededor de la pubertad. El equilibrio también puede verse afectado.
La pérdida de audición en el síndrome de Usher se debe a una mutación genética (alteración) que afecta a las células nerviosas de la cóclea, una estructura que transmite el sonido del oído interno. El mismo defecto genético también afecta negativamente las células fotorreceptoras en la retina, lo que lleva a la pérdida de la visión.
El Síndrome de Usher se transmite de los padres a su descendencia a través de un rasgo o condición de herencia autosómica recesiva. En este tipo de herencia, se requieren dos copias de un gen mutado [cambiado], uno de cada padre, para que el niño se vea afectado. Una persona con una sola copia del gen es un "portador" y rara vez tiene algún síntoma.
Los primeros síntomas de AMD suelen ser la distorsión visual de las líneas rectas y las dificultades en la lectura, reconocimiento de rostros y otras tareas visuales de foco fino. Muchos enfermos de AMD también son muy sensibles a la luz solar brillante. La visión lateral generalmente se conserva.
HAY DOS CLASES DE AMD: HÚMEDA Y SECO.
La diabetes es una enfermedad que ocurre cuando el páncreas no secreta suficiente insulina o el cuerpo no puede procesarla adecuadamente. La insulina es la hormona que regula el nivel de azúcar (glucosa) en la sangre.
El efecto de la diabetes en el ojo se llama retinopatía diabética. La RD es una complicación común de la diabetes, puede no tener ningún síntoma o no afectar la vista en las primeras etapas, pero a medida que avanza la enfermedad, la vista se verá afectada. Cuando la afección se detecta temprano, el tratamiento es efectivo para reducir o prevenir el daño a la vista.
Se cree que la RD es la forma más común de ceguera en personas en edad de trabajar en el mundo desarrollado. En alrededor del 10% de los casos, el edema macular diabético (EMD) puede ocurrir cuando los vasos sanguíneos pierden su contenido en la región macular de la retina y esto puede causar una forma más rápida de pérdida de visión.
La fase más temprana de la retinopatía diabética se conoce como "retinopatía diabética de fondo". A menudo no hay síntomas en las primeras etapas de la enfermedad, ni hay dolor. En esta fase, las arterias de la retina se debilitan y se filtran, formando pequeñas hemorragias. Estos vasos con fugas a menudo conducen a hinchazón o edema en la retina. A medida que la enfermedad progresa, algunos vasos sanguíneos que nutren la retina se bloquean, con el tiempo empeoran y privan a varias áreas de la retina con su suministro de sangre.
En la RD avanzada, las señales enviadas por la retina para la nutrición activan el crecimiento de nuevos vasos sanguíneos. Esta condición se llama retinopatía proliferativa. Estos nuevos vasos sanguíneos son anormales y frágiles. Crecen a lo largo de la retina y a lo largo de la superficie del gel transparente que llena el interior del ojo. Por sí mismos, estos vasos sanguíneos no causan síntomas ni pérdida de la visión. Sin embargo, tienen paredes delgadas y frágiles. Si pierden sangre, puede causar pérdida grave de la visión e incluso ceguera.
Una condición conocida como edema macular diabético ocurre cuando la sangre se filtra al centro de la retina, conocida como la mácula, la parte del ojo donde se produce una visión aguda y directa. El líquido hace que la mácula se hinche, borrando la visión. Esto puede ocurrir en cualquier etapa de la RD, aunque es más probable que ocurra a medida que la enfermedad progresa.
La prevención de la retinopatía diabética es el paso más importante para cualquier persona con diabetes. Los investigadores han encontrado que los pacientes diabéticos que son capaces de mantener los niveles adecuados de azúcar en la sangre y la presión arterial tienen menos problemas oculares que aquellos con un control deficiente. La dieta y el ejercicio desempeñan un papel importante en la salud general de las personas con diabetes.
Las personas con diabetes también pueden reducir en gran medida las posibilidades de complicaciones oculares acudiendo a exámenes regulares con un oculista.
La retinopatía diabética se trata de muchas maneras dependiendo de la etapa de la enfermedad y el problema específico que requiere atención. El médico se basa en varias pruebas para controlar la progresión de la enfermedad y tomar decisiones para el tratamiento apropiado. La cirugía ocular con láser llamada fotocoagulación pan-retina (PRP) es una opción de tratamiento para evitar que los vasos sanguíneos se filtren o para eliminar el crecimiento de vasos anormales y frágiles. Recientemente se ha disponible una nueva clase de medicamentos para tratar el edema macular y, a menudo, se usan junto con la terapia con láser. Estos son fármacos anti-VEGF (factor de crecimiento endotelial vascular) y se dirigen a la sustancia en el cuerpo que es responsable del desarrollo de los vasos sanguíneos. En el edema macular diabético, se produce demasiado VEGF en el ojo,
La vitrectomía es otra cirugía comúnmente necesaria para los pacientes diabéticos que sufren una hemorragia vítrea (sangrado en la sustancia similar al gel que llena el centro del ojo). Durante una vitrectomía, el cirujano de la retina extrae cuidadosamente la sangre, el tejido fibroso y el vítreo del ojo, lo que alivia la tracción sobre la retina y previene el desprendimiento de retina. Si se producen desprendimientos o desgarros de retina, a menudo se sellan con cirugía láser. El desprendimiento de retina requiere tratamiento quirúrgico para volver a unir la retina a la parte posterior del ojo. El pronóstico para la recuperación visual depende de la gravedad del desprendimiento.
La amaurosis congénita de Leber (LCA, por sus siglas en inglés) es una afección genética que causa pérdida grave de la visión en el nacimiento o en la primera infancia. El ACV causa ceguera infantil en una de cada 33,330 personas.
Síntomas
• Los síntomas a menudo se notan en las primeras semanas o meses después del nacimiento de un niño.
• Los padres pueden observar que el niño no enfoca sus ojos en las cosas de su entorno.
• Un niño puede exhibir movimientos oculares rápidos y "tambaleantes", llamados nistagmo. • Algunos niños pueden presionar o presionar sus ojos con sus dedos o puños.
Sinónimo de degeneración macular juvenil o distrofia macular pura.
enfermedad de Stargardt es una forma juvenil de la degeneración hereditaria de la mácula, que se caracteriza por manchas redondas de color amarillento o pisciformi alrededor de la mácula, en el nivel del epitelio pigmentario de la retina (RPE).
La enfermedad de Stargardt es la forma más común de distrofia hereditaria de la mácula, con una prevalencia de aproximadamente 1: 8,000-10,000.
La enfermedad ocurre característicamente en la primera o segunda década de la vida y se manifiesta con una reducción de la agudeza visual.
En las primeras etapas, la mácula generalmente muestra cambios obvios en el epitelio pigmentario de la retina, que son seguidos por la aparición de un área ovoidea horizontal de atrofia "de bronce".
En las etapas posteriores, las lesiones de la mácula se pueden asociar con una distrofia areolar central de la coroides. La angiografía con fluorescencia revela una característica oscura de la coroides ("silencio de la coroides"), probablemente debido a la acumulación de lipofuscina en el epitelio pigmentario de la retina. Esta enfermedad generalmente tiene una herencia autosómica recesiva, pero se han descrito algunas familias dominantes. La forma autosómica se debe a mutaciones en el gen ABCR, que codifica una proteína de transporte a través de la membrana celular, expresada en los segmentos externos de las varillas.
Se caracteriza por una historia de pérdida progresiva de la visión central y discriminación de color perjudicada, frecuentemente acompañada de fotofobia (aversión a la luz). Los hallazgos característicos del examen son: maculopatía en bull's eye y pérdida severa y selectiva de la función de los conos a la electrostina.
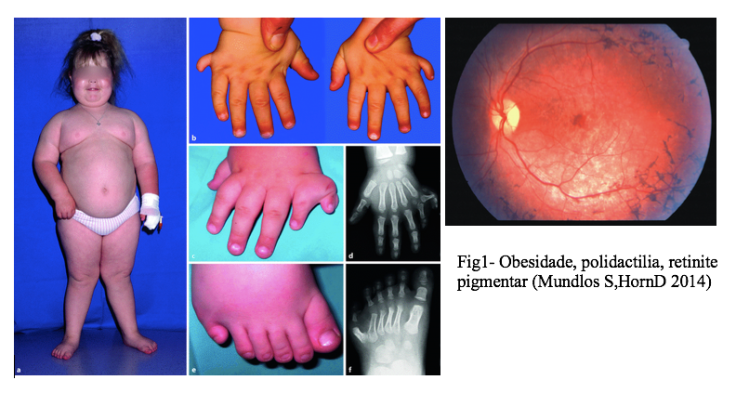
El síndrome de Bardet-Biedl es un trastorno hereditario raro que causa una serie de anomalías en el funcionamiento de ciertas partes del cuerpo, incluida la visión de una persona.
Síntomas
• Pérdida de la visión causada por la distrofia retiniana. La pérdida de la visión central a menudo se experimenta primero, con una pérdida de visión más severa a menudo experimentada por la adolescencia o la adultez temprana.
• Obesidad, generalmente más pronunciada en el área del torso (obesidad troncal).
• Dedos y / o dedos de los pies adicionales (polidactilia), o anormalidades menores de las manos y los pies, como pequeñas correas o piel extra entre los dedos de las manos y los pies.
• Carencia o disminución de características sexuales secundarias (hipogonadismo).
• Retraso en el desarrollo.
Es una enfermedad hereditaria poco frecuente. La persona es incapaz de absorber por completo las grasas de la dieta a través de los intestinos.
Abetalipoproteinemia; Acantocitosis; Deficiencia de apolipoproteína B
El síndrome de Bassen-Kornzweig es causado por un defecto en un gen que le ordena al cuerpo producir lipoproteínas (moléculas de grasa combinadas con proteína). El defecto le dificulta al cuerpo digerir apropiadamente grasa y vitaminas esenciales.
Los síntomas incluyen:
Puede haber daño a la retina del ojo RP ..
Los exámenes que se pueden hacer para ayudar a diagnosticar esta afección incluyen:
Puede haber disponibilidad de pruebas genéticas para las mutaciones en el gen MTP.
El tratamiento involucra grandes dosis de suplementos vitamínicos que contengan vitaminas liposolubles (vitamina A, vitamina D, vitamina E y vitamina K).
Igualmente, se recomiendan suplementos de ácido linoleico.
Las personas con esta afección deben hablar con un nutricionista. Se necesitan cambios en la alimentación para prevenir problemas estomacales. Esto puede implicar limitar la ingesta de algunos tipos de grasa.
Los suplementos de triglicéridos de cadena media se toman bajo la supervisión de un proveedor de atención médica. Se deben usar con precaución, debido a que pueden ocasionar daño hepático.
El pronóstico para la persona depende de la cantidad de problemas del sistema nervioso y el cerebro.
Las complicaciones pueden incluir:
Llame a su médico si su bebé o niño presenta síntomas de esta enfermedad. La asesoría genética le puede ayudar a las familias a entender la enfermedad y los riesgos de heredarla y aprender cómo cuidar de la persona.
Las dosis altas de vitaminas liposolubles pueden reducir el progreso de algunos problemas como el daño a la retina y la disminución de la visión.
Kliegman RM, St Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Defects in the metabolism in lipids. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 104.
Shamir R. Disorders of malabsorption. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21st ed. Philadelphia, PA: Elsevier; 2020:chap 364.
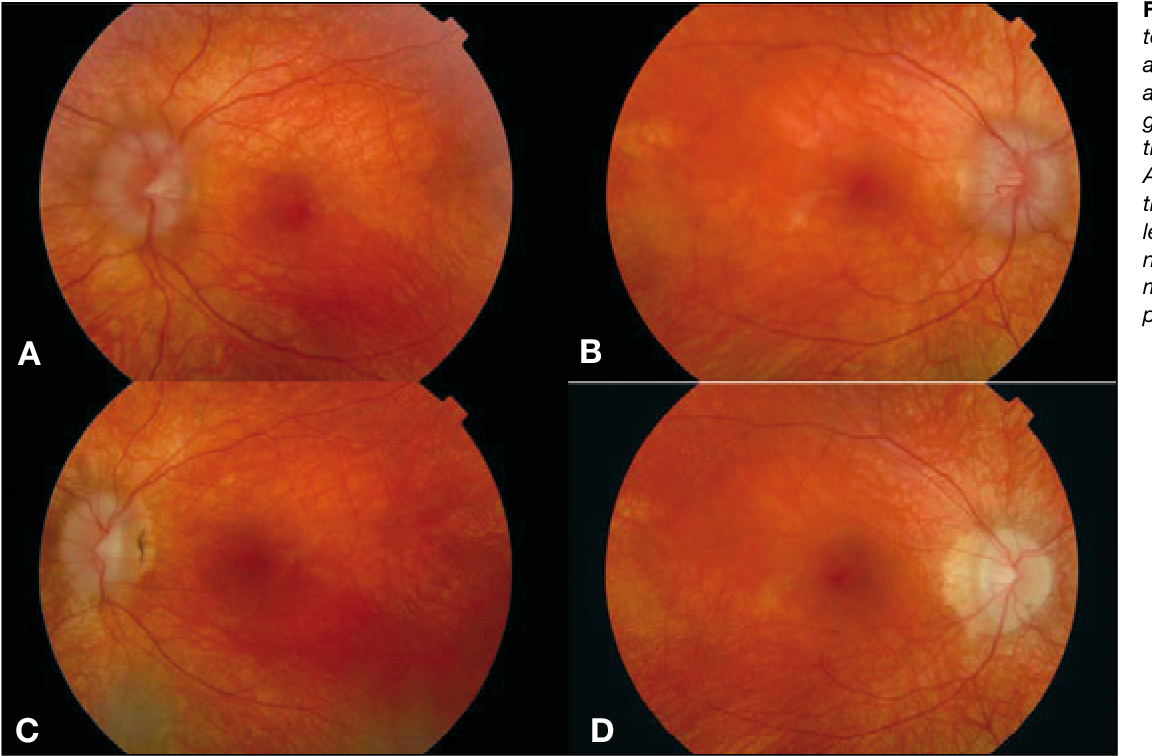
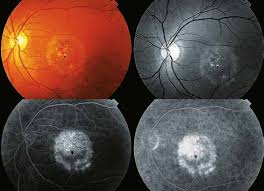
La enfermedad de Best (también conocida como distrofia macular viteliforme) es una forma hereditaria de degeneración macular caracterizada por una reducción de la visión central.
DESCRIPCIÓN CLÍNICA
La enfermedad de Best afecta la mácula, la parte central de la retina responsable de los detalles visuales finos y la percepción del color. La retina y sus células fotorreceptoras componentes son esenciales para la visión, ya que convierten la luz en impulsos eléctricos y luego transfieren estos impulsos al cerebro a través del nervio óptico.
Aunque la edad de inicio de enfermedad de Best puede variar, por lo general se diagnostica durante la infancia o la adolescencia. En las etapas iniciales, se forma un quiste amarillo brillante (saco lleno de líquido) en el epitelio pigmentario de la retina (EPR) debajo de la mácula. En el examen, el quiste se ve como un huevo soleado boca arriba. A pesar de la presencia del quiste, la agudeza visual puede permanecer normal o casi normal (entre 6/9 y 6/18) durante muchos años. La visión periférica (lateral, superior e inferior) generalmente no se ve afectada.
En muchas personas con la enfermedad Best, el quiste finalmente se rompe. Los depósitos de líquido y amarillo del quiste roto se extendieron por toda la mácula. En este punto, la mácula tiene una apariencia de huevo revuelto. Una vez que el quiste se rompe, la mácula y el RPE subyacente comienzan a atrofiarse, causando una mayor pérdida de visión. Como resultado, la visión central tiende a deteriorarse a aproximadamente 6/36 en la última etapa de la vida. Sin embargo, la enfermedad de Best no siempre afecta a ambos ojos por igual. Muchos individuos retienen una visión central útil en un ojo con una agudeza visual de alrededor de 6/12 en el ojo menos afectado.
En algunos casos, la enfermedad de Best no progresa lo suficiente como para causar una pérdida significativa de la visión central. Sin embargo, los especialistas en retina aún pueden detectar la enfermedad utilizando pruebas de diagnóstico sofisticadas que miden la función del RPE y la retina. Las personas con la enfermedad de Best también son a menudo hipermétropes y pueden corregirse con anteojos.
HERENCIA
La enfermedad de Best es transmitida genéticamente a través de las familias por el patrón de herencia autosómico dominante.
Laurence Moon es una afección más rara, pero similar a la de Bardet Biedel con cambios pigmentarios de la retina más cambios neurológicos progresivos y dificultades de aprendizaje.
La polidactilia no está presente.
Laurence Moon y B
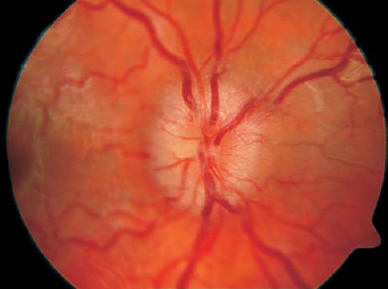
La coroideremia o "coroide denudata" es una distrofia coroidal-retinal rara; su curso es similar al de la distrofia de los conos y los bastones. Afecta, por regla general, a las personas masculinas.
Curso
Los primeros síntomas ocurren casi siempre antes de la edad de cuarenta. En los bordes de la periferia media del campo de visión, se forman scotomata (zonas defectuosas) que avanzan tanto hacia el centro como hacia el exterior, lo que causa una limitación cada vez mayor del campo visual. El curso de la coroideremia puede variar mucho de una persona a otra, y con frecuencia termina en ceguera.
La sensibilidad al deslumbramiento es alta y la adaptación a los cambios en el brillo gradualmente se vuelve más difícil.
A todo esto se agregan los problemas de visión en la oscuridad que pueden conducir a la ceguera nocturna.
A menudo, la agudeza visual central es bastante buena hasta la etapa final de la enfermedad. Incluso la visión del color (sentido cromático) permanece casi siempre intacta, los problemas ocurren solo en la enfermedad avanzada, en particular con las alteraciones de la percepción del azul.
En el segmento anterior del ojo solo se detecta una ligera miopía, que es típica de la coroidemia. En la parte inferior del ojo, en la periferia media y extrema, se encuentran áreas con una importante atrofia de la coroides y el epitelio pigmentario de la retina (EPR).
Con la evolución posterior de la enfermedad, estas áreas, previamente no claramente delimitadas, se vuelven más grandes y fluyen una hacia la otra para cubrir toda la retina.
En la retina, en lugar de depósitos típicos en forma de "corpúsculos óseos" (osteoplastos), podemos observar depósitos difusos bastante pequeños (pigmentaciones).
Durante mucho tiempo no se notan modificaciones de los vasos de la papila del nervio óptico. La representación de vasos sanguíneos en el fondo del ojo mediante fluorangiografía indica cambios en los vasos coroideos.
Este signo es importante para distinguir la coroideremia de otra enfermedad de la retina, la denominada retinosis pigmentaria –cromosóma X.
En el electroretinograma de campo completo (ERG), tanto las respuestas a los estímulos relacionados con la barra (ERG escotópica) como las relativas a los conos (ERG fotópico) ya están pronto por debajo de la norma. El examen de la mácula con el electrorretinograma multifocal (ERGmf) indica una reducción temprana en la respuesta de los conos.
La disminución en la respuesta electroretiniana evoluciona hacia adentro y hacia afuera hasta que la última etapa de la enfermedad tiene una respuesta de cono solo en la parte central de la mácula, siempre que haya una respuesta. El electrooculograma (EOG) ya está muy modificado, si no se extingue, muy pronto.
La coroideremia se debe a un defecto genético. La transmisión hereditaria es –cromosoma X . La mutación en un gen (CHM) en el cromosoma X causa la falta de una proteína específica (REP-1), que juega un papel importante en el epitelio pigmentario de la retina. El defecto genético causa la atrofia del epitelio pigmentado y la coroides. En la transmisión hereditaria del cromosoma X, la herencia relacionada con el sexo, como regla, solo los varones se ven afectados por la enfermedad.
Por lo general, las mujeres no se enferman ni tienen alteraciones visuales menores, aunque los depósitos de pequeñas manchas en el epitelio pigmentado son visibles en el examen de la vista.
Como portadores de un gen modificado (conductivo), pueden transmitir coroideremia a sus descendientes.
Los hijos varones de un presentador tienen entonces un 50 por ciento de posibilidades de contraer la coroideremia, mientras que para las hijas, la probabilidad de ser conductores es del 50 por ciento.
Sin embargo, no tendrán problemas de visión y este es un aspecto característico de la coroideremia. Con el fin de distinguir el choroideremia de una retinosis pigmentaria cromosoma X es importante saber que en la retina del corioideremia conductor se ve casi siempre pigmentaciones, mientras que este fenómeno es muy raro en conductora retinosis pigmentaria cromosoma X.
Para el diagnóstico diferencial, es importante también visitar a las madres de los niños afectados. Los hijos de un hombre con una corioiremia no contraen la enfermedad, mientras que todas las hijas son conductoras.
Métodos de diagnóstico
Además del procedimiento de diagnóstico descrito, los hombres con presunta corioidemia también pueden someterse a un examen genético-molecular o una prueba de anticuerpos específicos para determinar la expresión genética del gen REP1.
La enfermedad de Refsum es un trastorno extremadamente raro y complejo que afecta muchas partes del cuerpo. Una forma de la enfermedad degenerativa de la retina conocida como retinosis pigmentaria (RP) es una característica común de esta enfermedad.
La distrofia del cono y bastón es el resultado de una pérdida primaria de los fotorreceptores de los bastones , seguida de la pérdida de conos.
Las características son congénitas, hemeralopia estática y difusa coloración amarilla o gris del fondo. Después de 2 o 3 horas en total oscuridad, regresa el color normal del fondo. La condición es más frecuente en japonés. Ver hemeralopia (310500) para un comentario sobre el uso de este término, a diferencia del término nyctalopia.
La distrofia de la retina cono-vara (CRD, por sus siglas en inglés), característicamente conduce a un deterioro temprano de la visión. Una pérdida inicial de visión de color y de agudeza visual es seguida por nyctalopia (ceguera nocturna) y pérdida de campos visuales periféricos. En casos extremos, estos síntomas progresivos se acompañan de una pigmentación retina progresiva generalizada y una atrofia coriorretiniana de la retina central y periférica (Moore, 1992). Evans y col. (1995) encontraron ceguera total (sin percepción de la luz) en solo 3 de los 34 pacientes estudiados, y estos 3 tenían más de 65 años de edad. Efectos graves sobre la agudeza visual (percepción de la luz solamente) estuvieron presentes en otros 10 pacientes; sin embargo, su edad promedio fue de 60.3 años. Todos los demás pacientes conservaron algo de agudeza visual. En muchas familias, quizás la mayoría, no se encuentra atrofia de la atrofia coreorretiniana central y periférica (Tzekov,
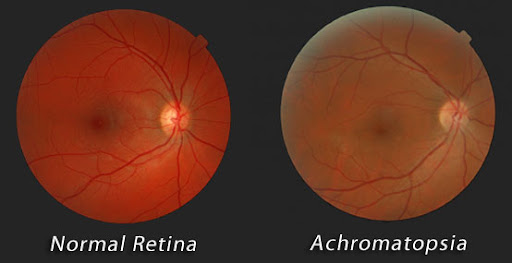
La acromatopsia es una condición hereditaria de la retina que causa una sensibilidad extrema a la luz (es decir, ceguera diurna), así como una disminución de la agudeza visual y la discriminación del color. La acromatopsia es causada por mutaciones en cualquiera de varios genes. Los genes más comunes asociados con la condición son los genes CNGB3 y CNGA3; las mutaciones en estos causan alrededor del 75 por ciento de los casos.
VISIÓN GENERAL
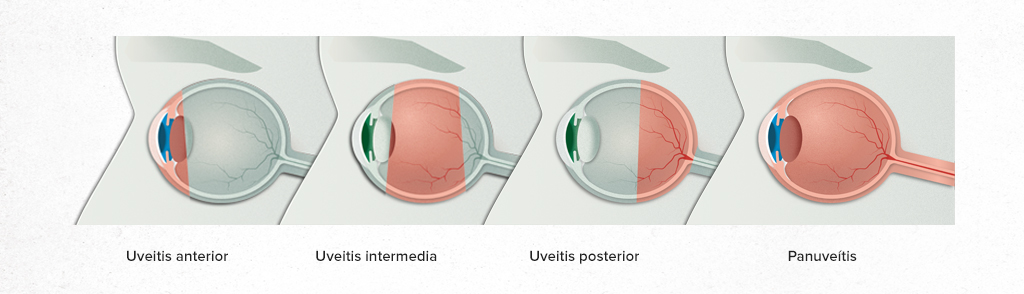
La uveítis es una enfermedad inflamatoria que daña el ojo. Afecta a diferentes partes del ojo, incluido el cristalino, la retina, el nervio óptico, el vítreo y la úvea, que incluye el iris, el cuerpo ciliar y la coroides. Afecta principalmente a personas de entre 20 y 50 años, pero puede ocurrir a cualquier edad. Es una de las principales causas de pérdida de la visión en adultos jóvenes y causa aproximadamente el 20 por ciento de la ceguera legal.
Cada año, alrededor del 2 por ciento de la población recibe un nuevo diagnóstico de uveíhttps://kit.fontawesome.com/08c5937065.jstis, una enfermedad que adopta diversas formas. Puede ser infeccioso o no infeccioso. Su oftalmólogo le ofrecerá un diagnóstico más específico, dependiendo de dónde se presente la enfermedad en el ojo.